Documentation
The core computation pipeline can separated in three major steps.
-
The pipeline collects the amino acid sequences of the proteins corresponding to the list of given identifiers.
-
The pipeline searches for resolved 3D structures of the respective proteins. In this step, the structures of
homologous proteins are also considered. A BLAST search of the up-to-date PDB instance is performed with the
E-value threshold of 10-10.
-
The spatial location of the amino acid residues affected by the mutations is analysed: The distances to all
other protein and nucleic acid (DNA and RNA) chains and hetero atom groups co-crystallyzed with each of the
identified homologs. This is called structural annotation. The common buffer components are ignored.
The distances, the similarity between the query and the structurally resolved proteins, and the quality of the
corresponding structure are combined into an Interaction Score that describes how likely
the nsSNP is to disrupt an interaction.
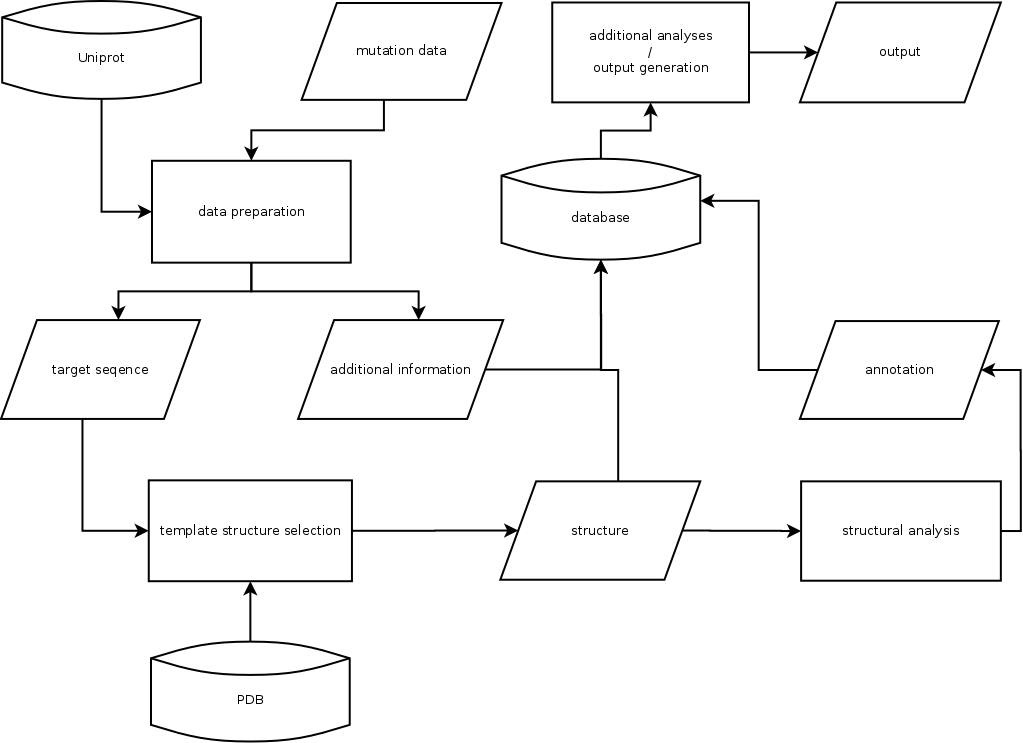
All the distances, contacts and scores are stored in a MySQL database. The database connects the produced
annotations with the input and all informations gathered on the way in order to produce outputs generated by
different additional analysis options.
Citation
Alexander Gress, Vasily Ramensky, Joachim Büch, Andreas Keller, and Olga V. Kalinina
StructMAn: annotation of single-nucleotide polymorphisms in the structural context
Nucleic Acids Research, first published online May 5, 2016
doi:10.1093/nar/gkw364
Tutorial
This is a simple tutorial going step-by-step through the most basic functions of StructMAn.
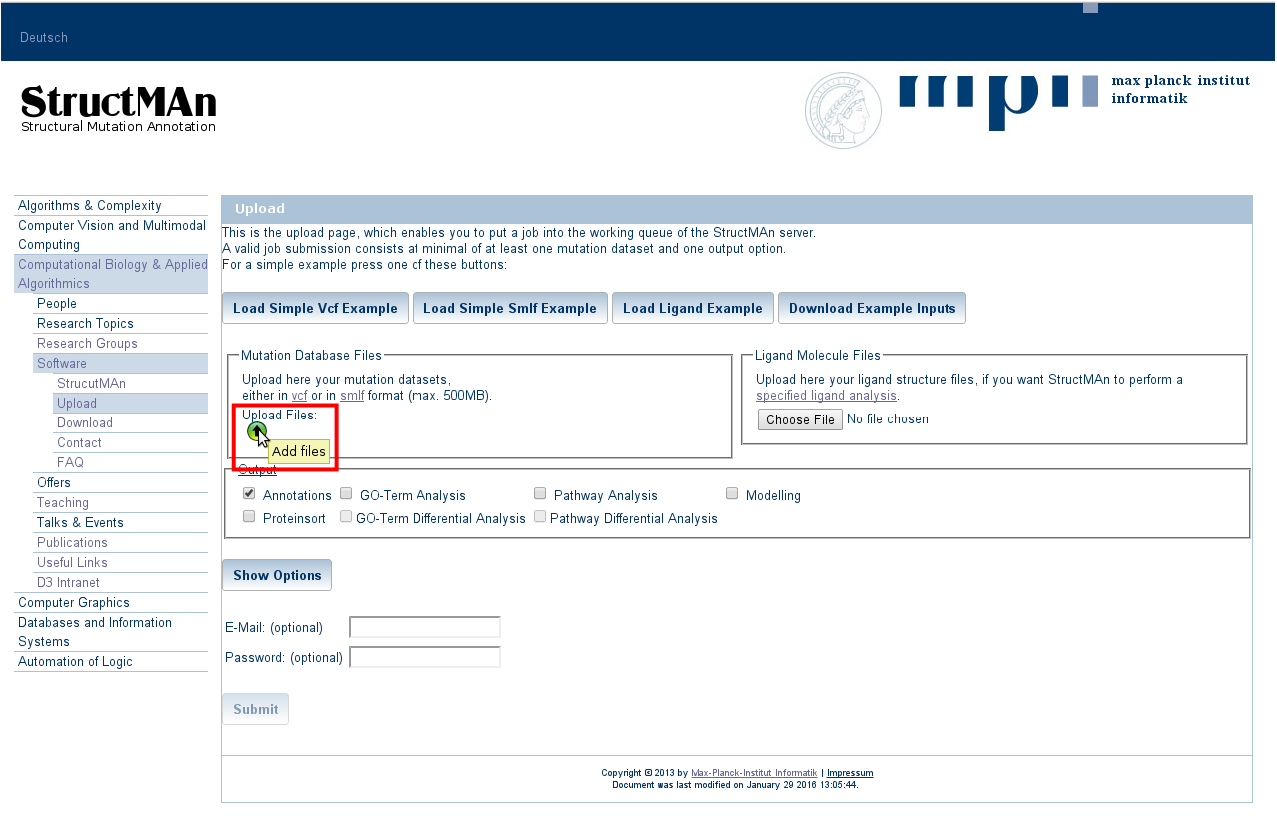
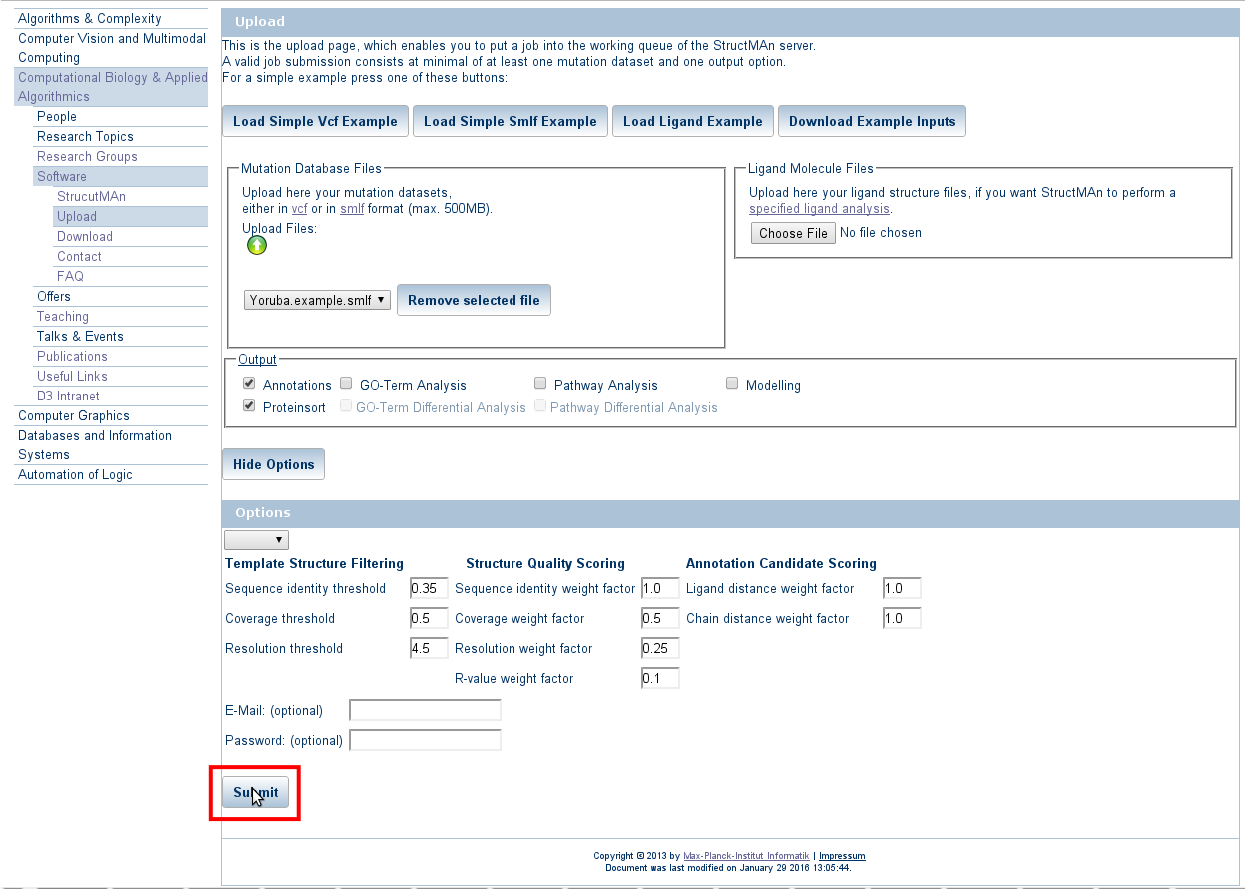
1. Uploading an input file
The simplest input file for StructMAn contains only information on mutations that need to be structurally studied.
Press on the green arrow button as shown in the picture below a file from your system. For bigger files, the upload may
take some time, especially for bigger vcf-files. The format of the uploaded file is checked and you will get an immediate
response, if the file format was not correct.
Mutation lists can be uploaded in the standard
VCF format
or a custom
SMLF format.
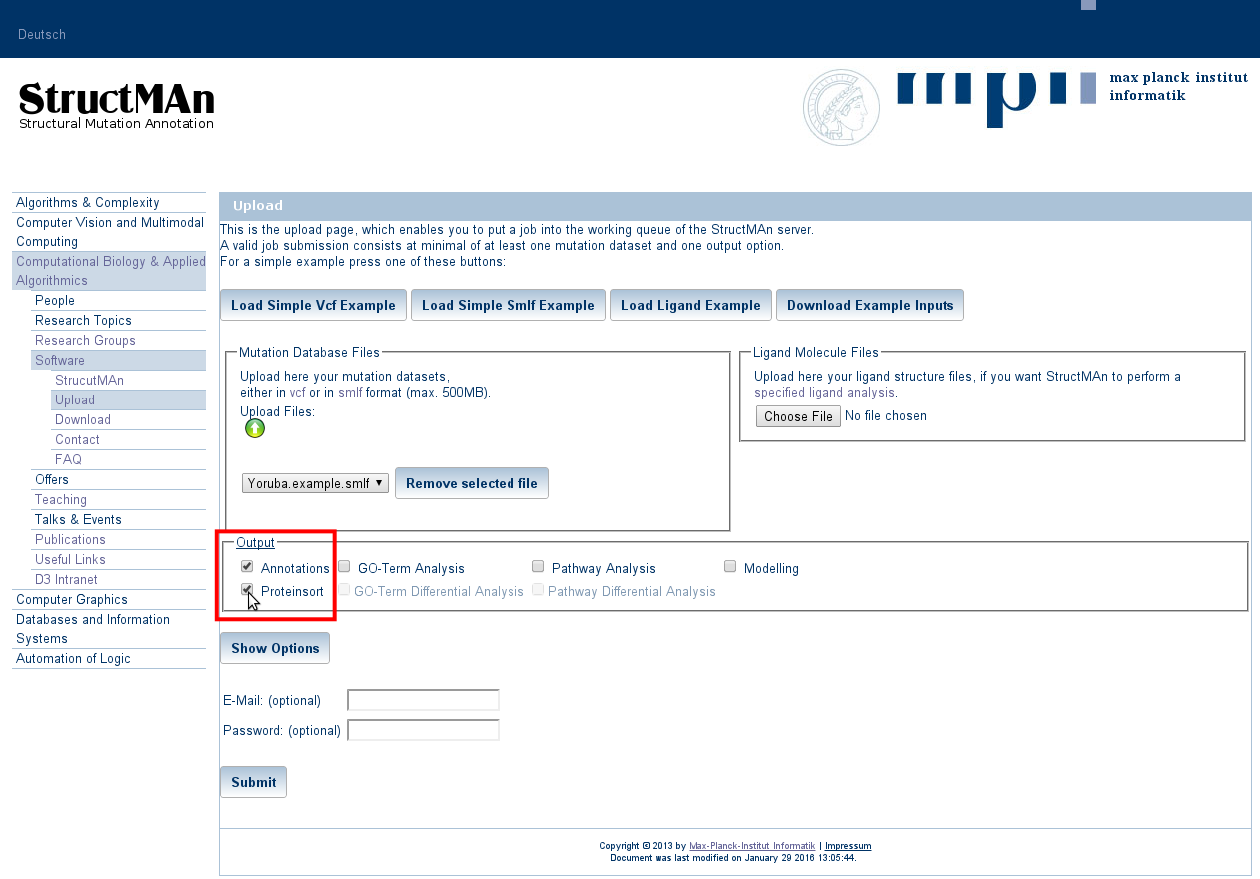
2. Choosing output options
The results are presented in a form of a table sorted either by the
mutation interaction score
or the
protein score. The order can be switched to protein
scores by selecting the "Annotations" and "Proteinsort" options. Selecting "Annotations" alone will result in
ordering by interaction score.
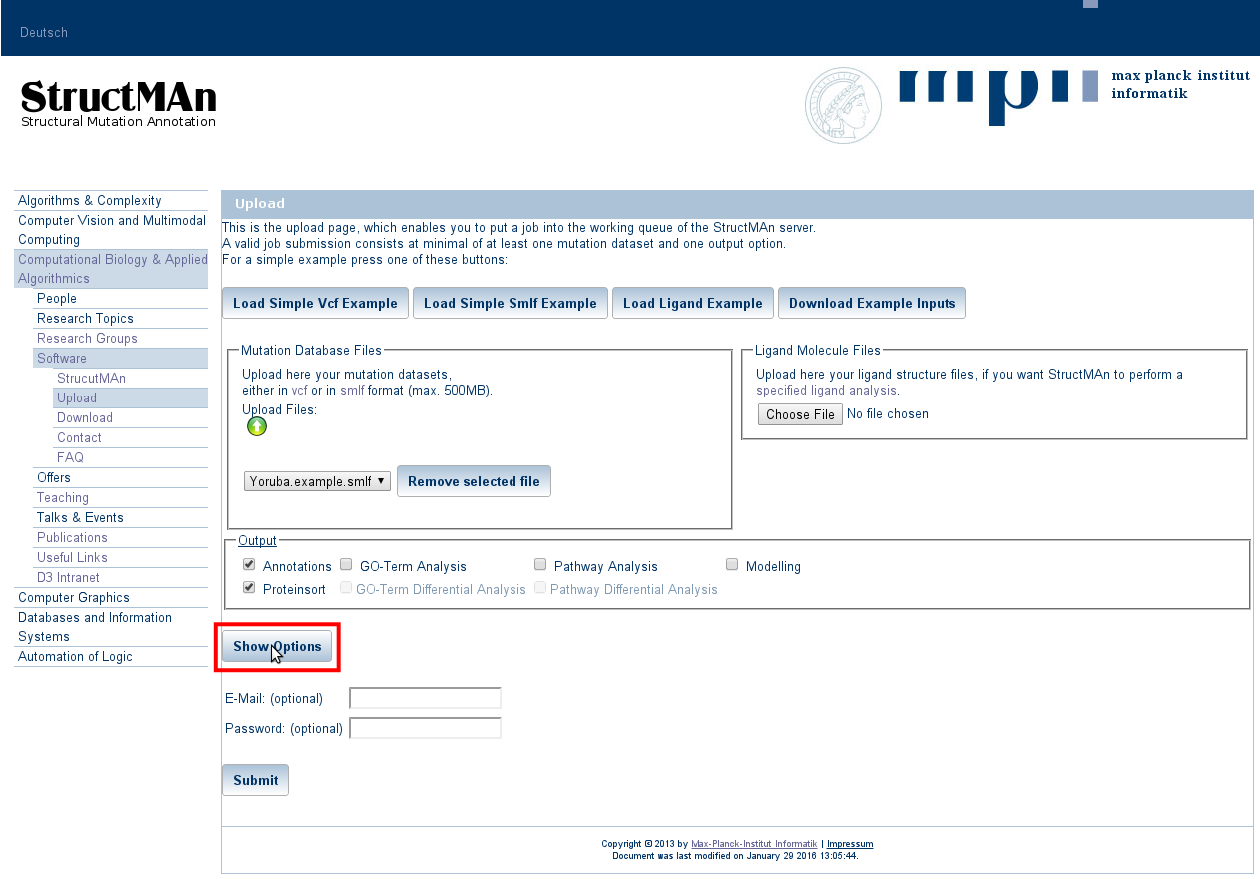
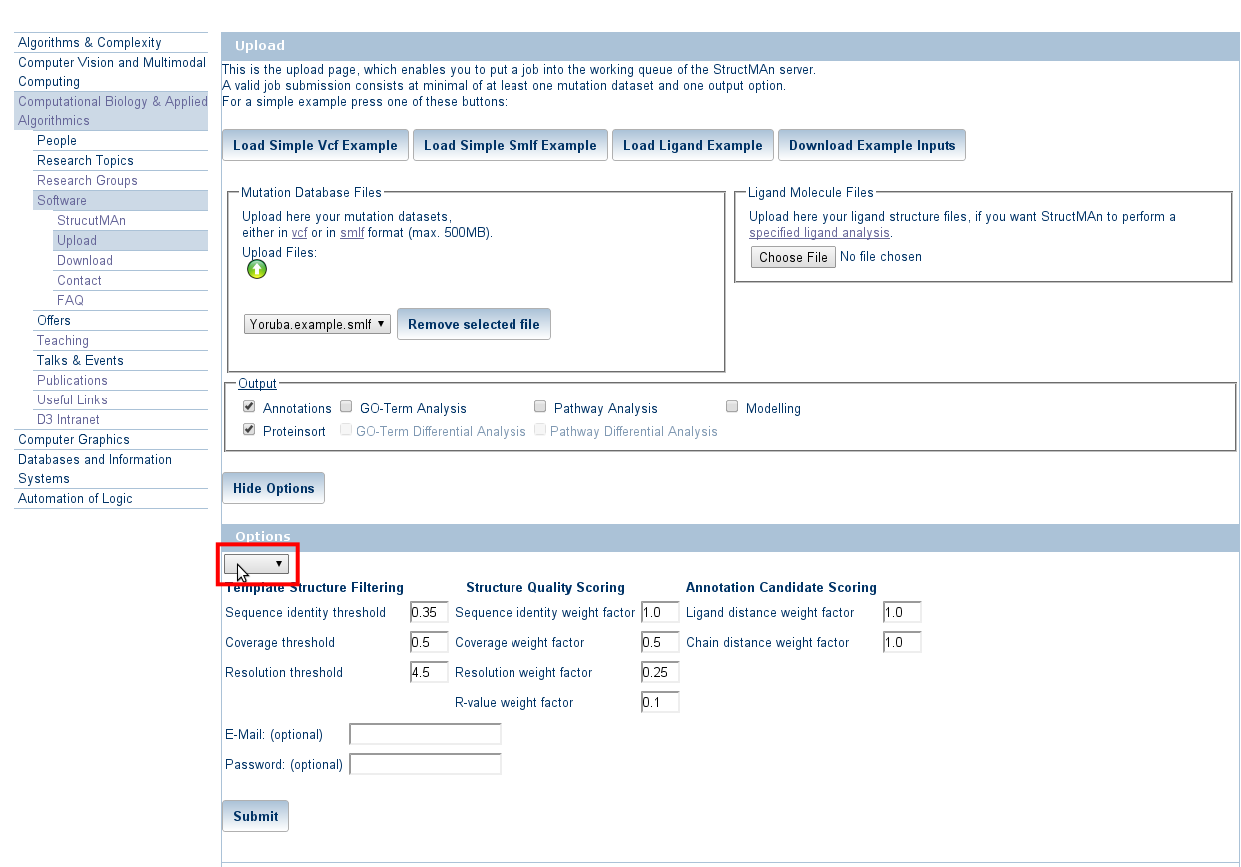
3. Expert Options
Press the "Show options" button to expand the expert options window.
Expert options allow to control the thresholds set for similarity search, structure selection and scoring.
For this basic tutorial choose "default". If you do not provide any expert options,
the server will also use these default values.
4. Submit your query
Press the "Submit" button.
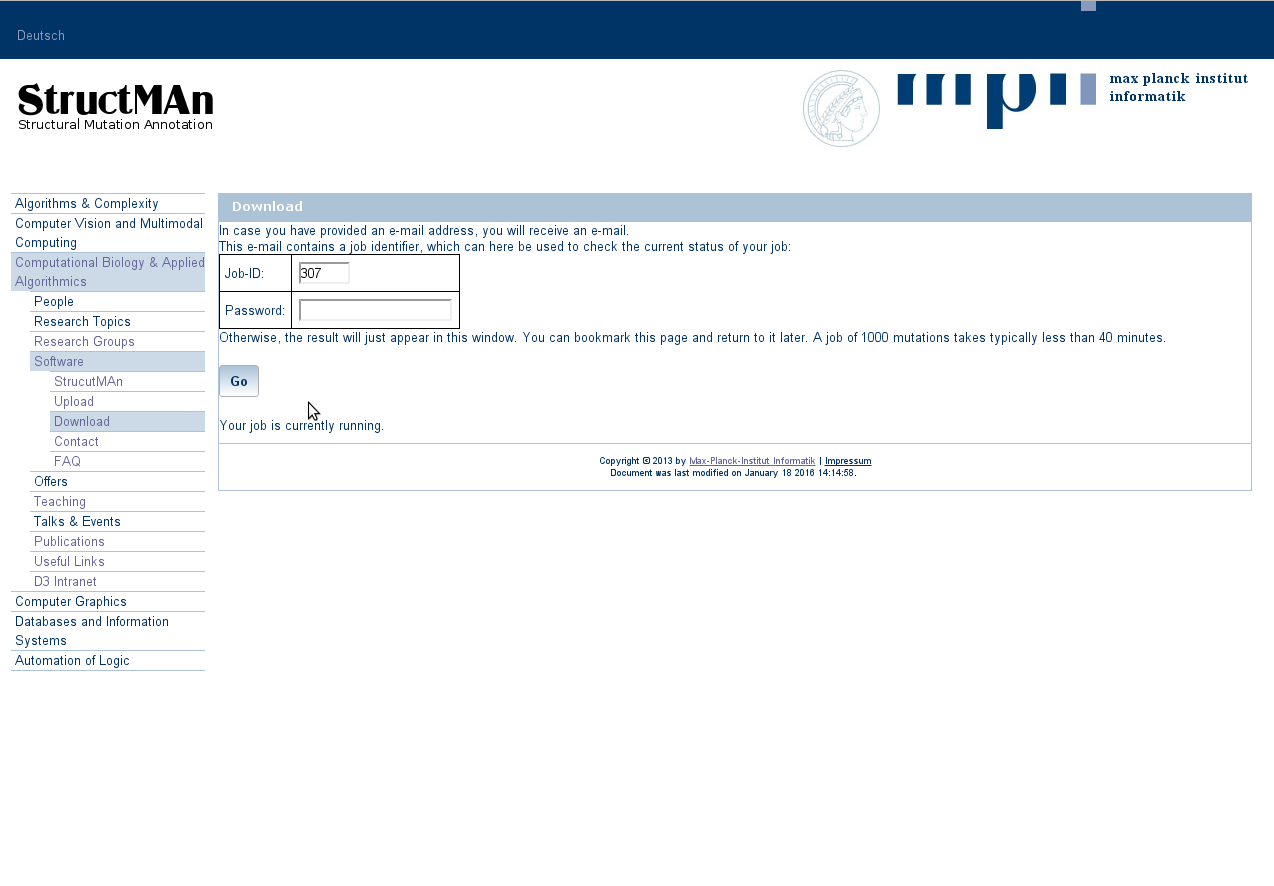
5. Wait for the results
After pressing the submit button, the server puts your job submission into a working queue and redirects you to
your personal results page. You can bookmark this page and come back to it later. The page shows you how many
jobs are in the queue before your job.
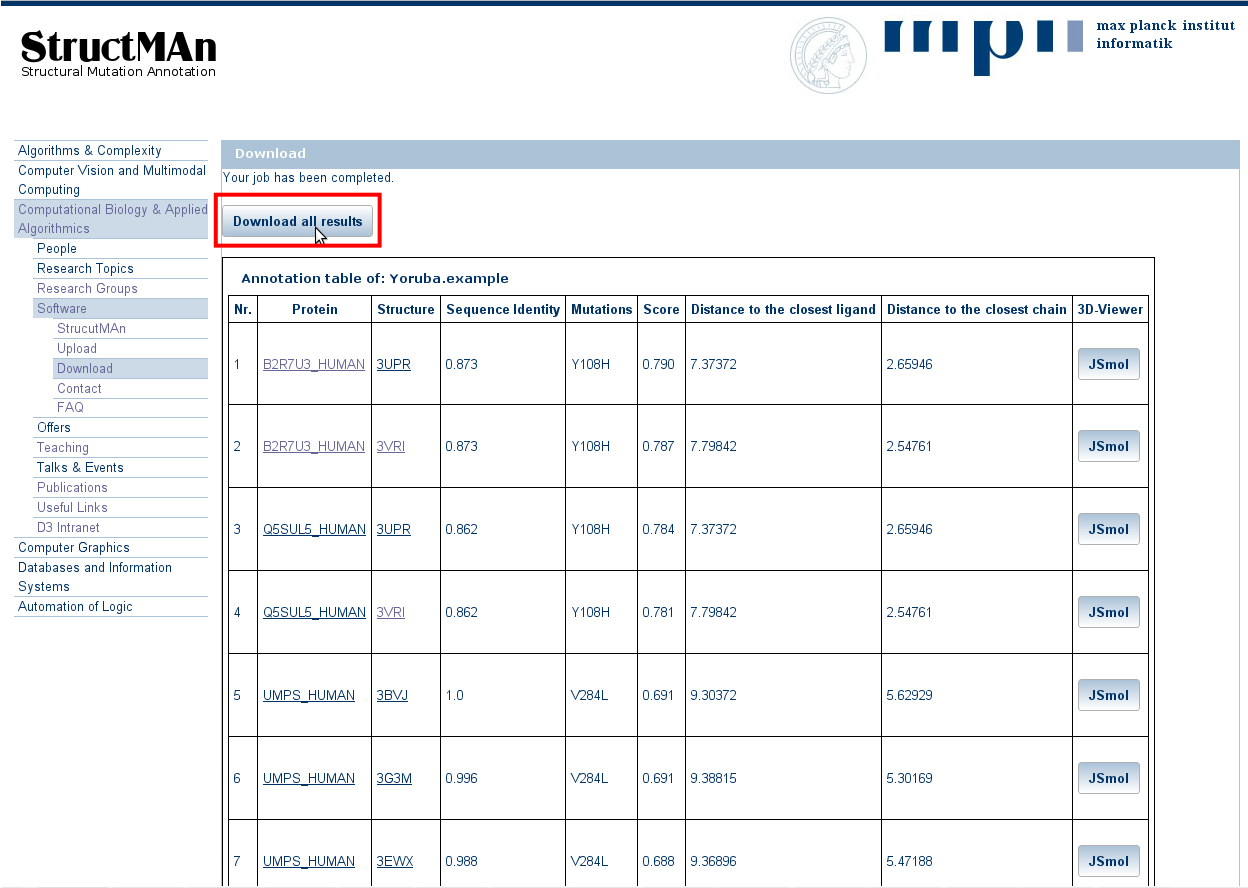
6. Viewing and downloading results
The results come as an interactive table, but are also downloadable via the "Download all results" button.
In the table, each row corresponds to a single mutation that can be mapped to a 3D structure of the same protein or a homolog.
The results are ordered using the
interaction score
or the
protein score.
7. Changing query options
If you want to change query options or the output mode, you have to resubmit your job, but it will run
virtually instantaneous using the produced data of the first job.
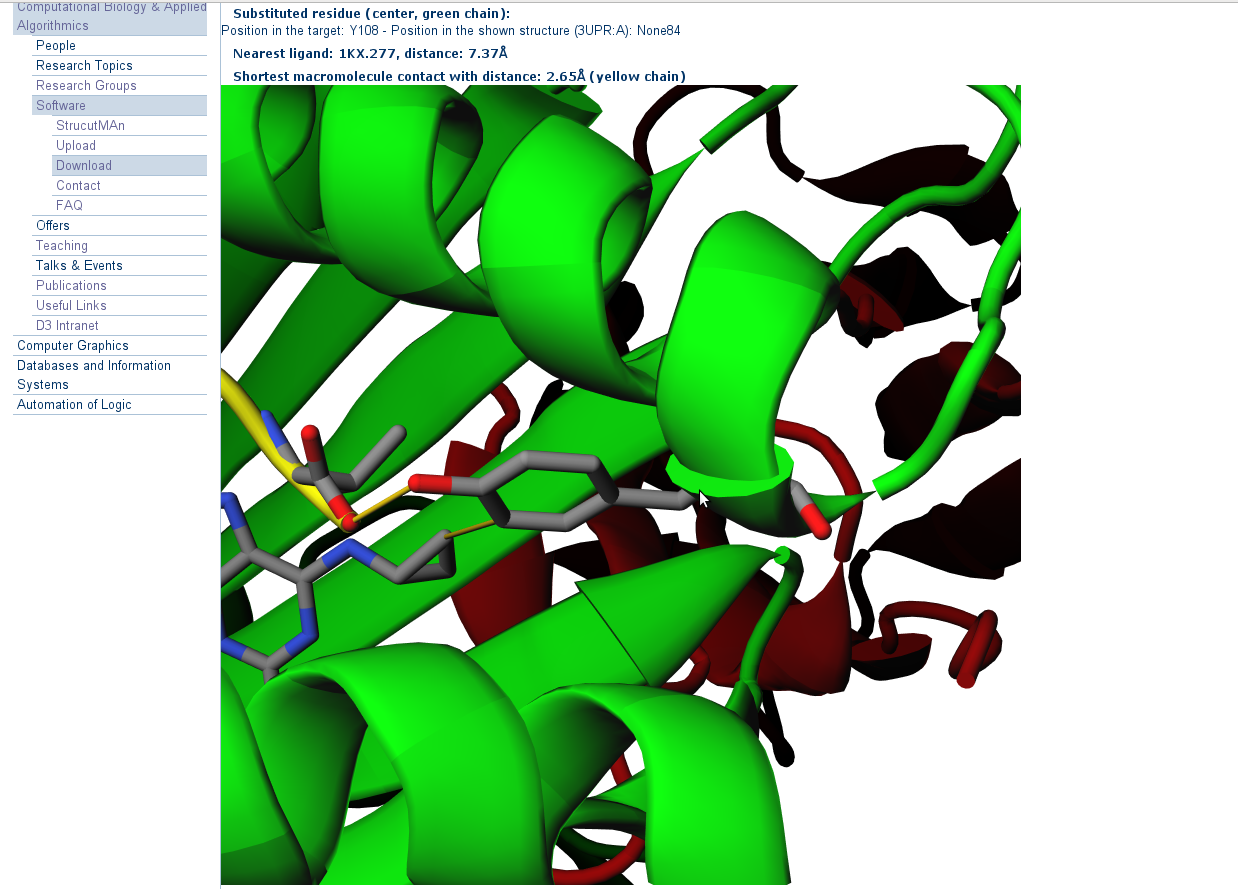
8. 3D-visualization of the mutations
Press on any "JSmol"-Button to open a new tab in browser showing the 3D-visualization of the mutation from the corresponding
table row. This may take some time to load depending on the size of the displayed structure. The initially displayed snapshot
is centered around the residue, which corresponds to the substituted one.